


Ferroptosis, an article published in the journal Cell gives you an insight into this hot research area

Brent R. Stockwell, the author of the origin of Ferroptosis, has written a review about research progress for the 10th anniversary of Ferroptosis, published in the journal Cell with a high impact factor of 66.85 (2022). The National Nature Foundation projects and the amount of money winning the bid in this direction are rising year by year, and the relevant research papers are also repeatedly topping the CNS! (Figure 1)

Figure 1. Key milestones and growth in the iron death literature over time
Ferroptosis is an iron-dependent lipid peroxidation form of cell death. This can be assessed in cell culture by testing whether iron chelators and lipophilic antioxidants inhibit cell death.
I. Three major ferroptosis research areas plate foundation—metabolic mechanisms, control of reactive oxygen species, iron regulation.
1. Amino acid and lipid metabolism mechanisms provide the basis for ferroptosis.
Reduced oxidized cystine leads to glutathione (GSH)-dependent cell death and can be prevented by vitamin E treatment. Phospholipids (PLs), a key component of membranes, when present in membrane lipids, the PUFA portion can act as a necessary peroxide substrate for ferroptosis.
2. The researchers elucidated the biological significance of oxidative damage of biomolecules.
System Xc- acts as an anti-transporter of input cystine, a key amino acid component of GSH, which prevents "oxidative stress" caused by oxidants such as hydrogen peroxide. Glutathione peroxidase 4 (GPX4), which is a selenium protein, GPX4 is a central inhibitor of ferroptosis and acts as a GSH-dependent peroxidase to combat oxidation of lipids in membranes. Erastin induces ferroptosis by blocking System Xc- uptake of cystine, leading to cys and GSH depletion, and the molecular target of RSL3 is GPX4, controlling a non-apoptotic form of oxidative cell death.
3. The importance of iron and iron regulation.
Fenton reported that iron salts reacted with peroxides to form hydroxyl radicals, proposing the reaction that now bears his name (Fe 2+ + HOOH → Fe 3+ + OH - + OH⋅). The main mechanism of iron transport in the form of transferrin (Tf). Together with the discovery of the iron export protein ferroportin, these studies have laid the foundation for our understanding of iron homeostasis.
Ⅱ. Ferroptosis mechanism

Figure 2. Mechanisms of ferroptosis
● Inactivation of acyl coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) rendered these cells resistant to two different GPX4 inhibitors - RSL3 and ML162. Overexpression of ACSL4 is sensitive to ferroptosis. The ACSL enzyme family is essential for ferroptosis, and activation of fatty acids to CoA esters is a key regulatory step in ferroptosis. PUFA biosynthesis is itself A means of regulating susceptibility to ferroptosis: by activating adenosine monophosphate activated protein kinase (AMPK) to drive resistance to ferroptosis, and by controlling acetyl-CoA carboxylase (ACC) to restrict PUFA biosynthesis. In conclusion, PUFA-containing membrane-localized lipids are drivers of ferroptosis, including PL, ether lipids, and other glycerol-derived lipids.
● Monounsaturated fatty acids (MUFA), such as oleic acid, require ACSL3 for their anti-ferroptosis effects.
● The multidrug resistance gene MDR1 increases the sensitivity to ferroptosis by causing the loss of GSH, while the cys catabolic enzyme cys dioxygenase 1 (CDO1) increases the sensitivity to ferroptosis by breaking down cys and then consuming GSH.
● Compound FIN56 induces degradation of GPX4 and increases sensitivity to ferroptosis by overdepletion of coenzyme Q10 (CoQ10) in the mevalonate pathway.
● GPX4 independently controls lipid ROS accumulation. However, in the last few years, three systems for inhibiting ferroptosis that are not dependent on GPX4 have been identified. Ferroptosis suppressor protein 1 (FSP1)/CoQ 10, dihydroorotic acid dehydrogenase (DHODH) and GTP cyclization hydrolase 1 (GCH1)/tetrahydrobiopterin (BH4) inhibit ferroptosis independently of GPX4. Cells with high expression of GCH1 or DHODH were more resistant to ferroptosis, whereas cells with low expression were more sensitive to ferroptosis.
● The amino acid oxidase interleukin-4-induced-1 (IL4i1), originally identified in B cells as a gene responsive to IL-4 induction, produces the metabolite indole-3-pyruvate (In3Py), which inhibits the ferroptosis scavenging mechanism via free radicals and a gene expression profile coordinated to attenuate ferroptosis.
● Peroxidation of PUFA-containing lipids is driven by an intracellular iron pool, and active iron triggers the Fenton reaction, which initiates the formation of lipid peroxides that are substrates for the Fenton reaction by triggering lipid peroxidation as well as the production of iron-dependent enzymes such as arachidonic acid lipoxygenases (ALOXs). These lipid peroxides are substrates for the Fenton reaction. 15-Lipoxygenase can be complexed with PE-binding protein 1 (PEBP1) to specifically catalyze the enzyme's substrate from free PUFA to PUFA- PL. In addition, p53-dependent ferroptosis requires 12-lipoxygenase. Cytochrome P450 oxidoreductase (POR) also contributes to lipid peroxidation during ferroptosis, suggesting that several iron-containing enzymes have the ability to promote lipid peroxidation leading to ferroptosis.
● Other mechanisms that control the abundance of iron in the cell influence the cell's sensitivity to ferroptosis: Iron output by ferroportin or prominin-2 (prom2) -mediated ferritin polyvesicule (MVB) enhances the cell's resistance to ferroptosis, as this depletes the iron in the cell to reduce the ability of lipid peroxidation.
● Erastin and RSL3 represent the first two classes of iron-death-inducing compounds that acting by inhibit cystine uptake via System Xc- and GPX4, respectively. FIN56 induces the degradation of GPX4 as a third class compound, FINO2 represents the fourth class of ferroptosis-inducing compounds, and FINO2 oxidizes Fe(II) to Fe(III) : iron chelating agents more effectively inhibit its lethal activity. The wider distribution of oxidized lipids detected after FINO 2 treatment of cells than after treatment with erastin suggests that FINO2 reacts with Fe(II) in a Fenton reaction to produce alkoxyl radicals that directly initiate lipid peroxidation.
● Three major mechanisms of resistance to ferroptosis have been identified - the antioxidant regulator NRF2, the transsulfuration pathway and the mechanistic target of rapamycin (mTOR).
Ⅲ. Physiological function of ferroptosis

Figure 3. Physiological functions of ferroptosis
A key advance in the search for biological processes involved in ferroptosis has been the discovery of markers for detecting ferroptosis. Given that ferroptosis is driven by iron-dependent lipid peroxidation, detection of such lipid peroxidation events during ferroptosis is essential (Figure 4). In addition, mitochondria typically exhibit a shrunken, dense morphology during ferroptosis, and specific gene expression changes can be detected. TfR1 up-regulation and movement toward the plasma membrane mark iron-dead cells. Using these markers, many of the natural physiological functions of this ancient form of cell death have been discovered.
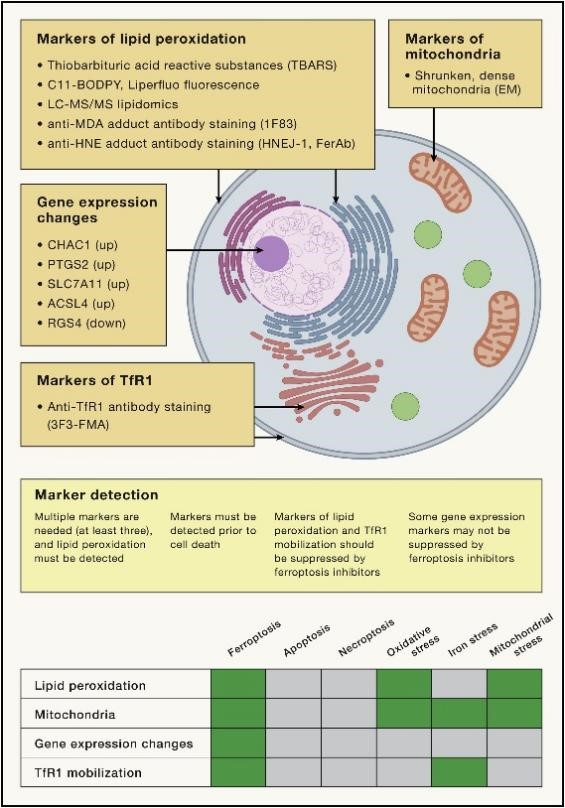
Figure 4. Markers of ferroptosis
Ⅳ. Pathologic background involving ferroptosis
In addition to the normal physiological functions of ferroptosis described above, ferroptosis is also associated with many pathologies (Figure 5).

Figure 5. Role of organelles and organs in ferroptosis
1. Iron overload disease
A recent report found that mice fed a high-iron diet and mice with mutations associated with inherited hemochromatosis developed hepatic damage in markers of ferroptosis, which was reversed by ferrostatin 1 inhibitors.
2. Organ damage
A recent study reports that multiple organ dysfunction syndrome (MODS) is common in critically ill patients in intensive care units (icus), and it involves ferroptosis. Plasma levels of catalyzed iron and malondialdehyde (a marker of lipid peroxidation) in 176 critically ill adult patients were positively correlated with patient Assessment of Sequential Organ Failure (SOFA) scores. In addition, administration of iron sulfate to mice resulted in multiorgan damage, raising markers of damage in kidney, liver, muscle, heart, and plasma, which was attenuated by inhibitors of ferroptosis (e.g., vitamin E). Thus, inhibiting ferroptosis may be a viable strategy for preventing multiple organ damage in an intensive care setting.
3. Retinal degeneration
Retinal pigment epithelial cells undergo degeneration and death during retinal diseases (e.g., age-related macular degeneration). Prussian blue nanoparticles can inhibit the death of these cells.
4. Neurodegeneration
Numerous reports have linked ferroptosis to a variety of neurodegenerative diseases, including Huntington's disease (HD), Alzheimer's disease (AD), PD, and amyotrophic lateral sclerosis (ALS). One preclinical and clinically effective ALS treatment, CuII(atsm), inhibits ferroptosis.
5. Infectious diseases
Hepatitis C is a major cause of liver disease, and hepatitis C virus replication is limited by activation of ferroptosis in host cells.
Severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) may activate ferroptosis during infection.
M. marinae infection in zebrafish is inhibited by heme oxygenase 1 (HMOX1), which regulates the availability of iron in heme through treatment with ferrostatin 1.
Ferroptosis enhances Pseudomonas aeruginosa infection.
6. Autoimmune diseases
Systemic lupus erythematosus (SLE) is an autoimmune disease associated with the activation of ferroptosis in neutrophils. Asthma, which involves over-activation of the immune response, may therefore be triggered or worsened by ferroptosis in airway epithelial cells that release immunogenic mitochondrial DNA. Ferroptosis, when it occurs in neutrophils or airway epithelial cells, leads to excessive immune activation, resulting in autoimmune disease.
7. Tumors
Although ferroptosis has been reported to function as a tumor suppressor mechanism, the loss of ferroptosis can drive tumorigenesis. Selenoproteins are predictors of cancer risk and selenium is elevated in tumor tissue, suggesting elevated GPX4 abundance and activity in tumorigenesis.
Ⅴ. Therapeutic applications of ferroptosis
Currently, monitoring lipid peroxidation is one way to recognize the presence of ferroptosis. Methods for detecting lipid peroxidation include the use of a thiobarbituric acid reactive substances (TBARS) assay for the detection of oxidized or lipid peroxidation products of isoprostane by LC-MS/MS, C11-BODIPY fluorescent probes, and antibodies reacting with antibodies detecting adducts formed by lipid peroxidation products such as the anti-HNE FerAb antibody, HNEJ-1 antibody and anti-malondialdehyde (MDA) adduct 1F83 antibody, 3F3-FMA antibody, and other anti-TfR1 antibodies for detection. Several genes, such as CHAC1, PTGS2, SLC7A11, and ACSL4, are induced during ferroptosis, while RGS4 is down-regulated during ferroptosis. The altered expression of these genes can be detected by qPCR as an indicator of ferroptosis.
1. Promoter of ferroptosis
As a way of cell death, ferroptosis could potentially be used to eliminate problematic cell types, such as cancer cells, inflammatory cells, or activated fibroblasts. Four broad mechanisms have been identified to induce ferroptosis: (1) inhibition of System Xc-, (2) inhibition/degradation/inactivation of GPX4, (3) depletion of reductive CoQ10, and (4) induction by lipids: peroxidation through overload of peroxides, iron, or polyunsaturated fatty acids.
- ● Inhibition of System Xc- is a potent mechanism for inducing ferroptosis - Multiple structurally distinct small-molecule inhibitors of this anti-translocator protein, such as erastin, sulfasalazine, and Glu, inhibit System Xc- and induce ferroptosis.
● Cysteine reduction removes extracellular substrates of System Xc- and also induces ferroptosis.
● Gene inactivation or small molecule-mediated inhibition or degradation of GPX4 induces ferroptosis in many cell types.
● Inhibition of CoQ10 biosynthesis via the mevalonate pathway and inactivation of CoQ10 reductases (e.g., AIFM2/FSP1 or DHODH) induces ferroptosis.
● Ferroptosis can be induced by treatment with excess iron, PUFA, or peroxides (such as tBOOH or FINO 2 ).
2. Ferroptosis inhibitors
● Ferrostatin-1 and liproxstatin as free radical trapping agents to inhibit the propagation of lipid peroxidation.
● The cholesterol-lowering drug probucol inhibits ferroptosis and is effective in a Glu toxicity model.
● Necrostatin-1 (nec-1) is a RIPK1 inhibitor that inhibits necroptosis.
● Selenium delivery inhibits ferroptosis during stroke.
● The mitochondrial-targeted nitric oxide XJB-5-131 inhibits apoptosis and ferroptosis.






 Chemical Structure")



-FINO2 Chemical Structure")




Kommentare