


Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis
Streptococcus pyogenes, also known as group A streptococcus (GAS), causes a wide variety of acute infections, ranging from localized purulent infections to severe, even fatal, invasive disease. Systemic spread is usually caused by bacterial penetration of the epithelial barrier of the pharynx or damaged skin and, if not well controlled, can lead to blood and soft tissue invasion. Superficial colonization and invasive infection of GAS depend on secreted GAS virulence factors, of which cysteine protease exotoxin (SpeB) is the key. SpeB is initially an inactive zymogen that is proteolytically converted to a mature catalytically active enzyme. SpeB contributes to epidermal localization and systemic spread, but the underlying mechanisms are unknown.
Gasdermins, encoded by five paralogous genes in humans (GSDMA, GSDMB, GSDMC, GSDMD, and GSDME), are a family of pore-forming proteins that act as keys to pyroptosis. After recognizing invasive bacteria or other danger signals, innate immune receptors form large supramolecular complexes called inflammasomes that activate inflammatory caspases, cleave GSDMD and release the N-terminus. GSDMD-NTs assemble in the cell membrane, forming pores and triggering pyroptosis. Individual gasdermins are selectively expressed at specific mucosal sites, with GSDMA predominantly expressed in the skin and epithelial cells of the upper gastrointestinal tract. But how GSDMA is activated remains unknown. Recently, Liu Xing's team published an article in Nature, reporting on the response mechanism of GAS infection.
SepB induces lytic death of epithelial cells in the skin:
To study the host cell response to GAS infection, mice were infected with the following different types of GAS strains: wild-type WT M1T1 strain, isogenic mutant strains (ΔcepA, Δmac or ΔspeB), and animal-passaged covR/S - strains. It was found that GAS WT, ΔcepA, and Δmac, which were normally expressed by SpeB, caused severe purulent and necrotic lesions at the site of skin infection, while GAS SpeB-deficient ΔspeB and covR/S-strains showed no obvious response (Fig. 1a,b). GAS WT, ΔcepA, and Δmac mutant-infected mice had disrupted epithelial integrity and increased neutrophil infiltration, whereas ΔspeB, covR/S-mutant-infected mice had tissue damage and neutrophils compared to the former. Infiltration was significantly reduced (Fig. 1c,d). But the latter had more severe systemic infections, significantly increased bacterial load in the spleen and liver, and were more likely to die. Reintroduction of SpeB into ΔspeB mutant-infected mice restored GAS-induced skin damage, increased neutrophil infiltration and reduced systemic infection caused by GAS ΔspeB infection, confirming the role of SpeB in GAS M1T1 infection.
To investigate how SepB exerts its function during GAS infection, the researchers isolated primary mouse keratinocytes and infected them with the GAS strain. It was found that GAS WT, ΔcepA, and Δmac infection resulted in massive keratinocyte death with typical pyroptotic balloon-like morphology and release of lactate dehydrogenase (LDH), while those infected with ΔspeB, covR/S-mutant strains did not. (Fig. 1e,f). To further evaluate the role of SpeB in cell death, recombinant SpeB and its catalytically inactive mutant C192S (mSpeB) were introduced into mouse primary keratinocytes by electroporation. Electroporation of WT SpeB efficiently resulted in cell death with the hallmarks of pyroptosis—the swelling of the cell membrane and release of LDH, which was not seen in mSpeB (Fig. 1g,h). Thus, these data suggest that SpeB is a potential trigger for lytic death of skin epithelial cells.

Figure 1 GAS SpeB induces lytic death of skin epithelial cells
SpeB triggers GSDMA-dependent pyroptosis
To verify the role of GSDMA, the researchers used CRISPR/Cas9-mediated gene editing to knock out GSDMA in A431 cells. Compared with SepB transfection-induced WT cells, GSDMA-depleted cells were highly resistant to SpeB-triggered lytic cell death (Fig. 2c,d). Meanwhile, neither GSDMA alone nor SpeB alone could alter the morphology and viability of 293T cells lacking endogenous GSDMA, whereas co-expression of GSDMA with SpeB resulted in pyroptosis with balloon-like morphology and massive LDH release (Fig. 2e,f). . Notably, co-transfection of mSepB and GSDMA did not result in cell death; the caspase inhibitor E64 completely abolished SpeB-induced pyroptosis, while the caspase inhibitor (Z-WEHD) -fmk, Z-DEVD-fmk and Z-VAD-fmk) no. Thus, catalytically active SpeB induces GSDMA-dependent but not caspase-independent pyroptosis.
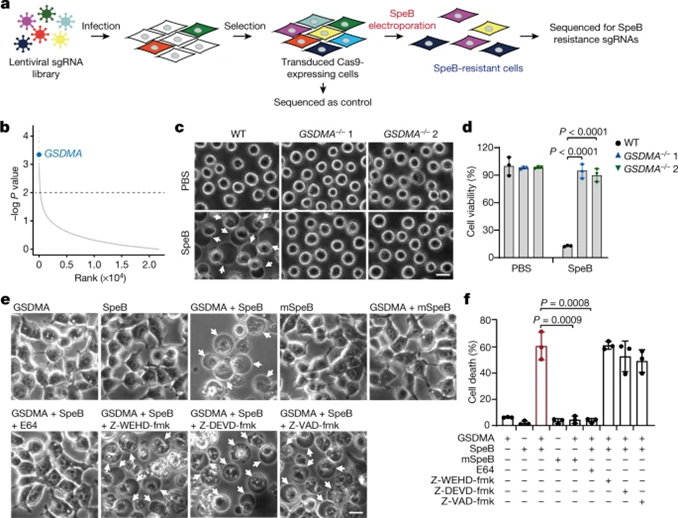
Figure 2 SpeB triggers pyroptosis in a GSDMA-dependent manner
SpeB cleaves GSDMA after Gln246
We also found that, in contrast to mSpeB, co-expression of GSDMA with WT SpeB resulted in a cleaved N-terminal fragment (GSDMA-NT) that was undetectable in the presence of the cysteinase inhibitor E64, but not in cysteine Detectable in the presence of aspartase inhibitors (Fig. 3a). In vitro cleavage experiments showed that treatment of recombinant GSDMA with WT SpeB in a dose-dependent manner, compared with mSpeB, produced the N-terminal p27 and C-terminal p23 fragments of GSDMA (Fig. 3b), in the presence of the cysteinase inhibitor E64 could not be detected (Fig. 3c). Co-expression of SpeB and gasdermins in 293T cells indicated that SpeB could specifically cleave GSDMA, but not GSDMB, GSDMC, GSDMD, and GSDME (Fig. 3d). GAS WT, ΔcepA, Δmac-infected A431 cells led to lysis of GSDMA, but not GSDMD, GSDME, and lysed gasdermins were not detected after infection with ΔspeB, covR/S-mutants (Fig. 3e). These data indicate that GSDMA exhibits specificity in response to SpeB and is not indiscriminately cleaved by other proteases.
The researchers also used Edman sequencing and found that SpeB cleaves GSDMA after Q246 in the GSDMA-linking region. To confirm I245/Q246 as the cleavage site, we utilized I245N, Q246E and I245N/Q246E GSDMA mutants and tested their sensitivity to SpeB (Fig. 3f,g). The I245N and I245N/Q246Q mutant GSDMA were completely resistant to SpeB-mediated cleavage, while the Q246E mutant remained cleaved. Thus, SpeB directly and selectively cleaves GSDMA in the junction region after Gln246.

Figure 3 SpeB cleaves GSDMA after Gln246
SpeB-cleaved GSDMA-NTs cause pyroptosis
To further explore the role of GSDMA cleavage in SpeB-induced pyroptosis, we introduced WT or catalytically dead SpeB into 293T cells expressing WT GSDMA or GSDMA (I245N/Q246E) without being cleaved by SpeB, and assessed GSDMA cleavage and cell pyroptosis. GSDMA cleavage, pyroptosis and LDH release occurred only in WT SpeB and WT GSDMA-transfected cells (Fig. 3h,i), indicating that SpeB-mediated GSDMA cleavage is necessary and sufficient for SpeB-induced pyroptosis. To determine the role of the SpeB cleavage product of GSDMA in SpeB-induced pyroptosis, we treated recombinant full-length GSDMA with SpeB and the purified GSDMA-NT and C-terminal (GSDMA-CT) reaction products were introduced into 293T by electroporation in cells. It was found that electroporation of purified GSDMA-NT or GSDMD and caspase-11 (positive control) resulted in severe pyroptosis, whereas electroporation of full-length GSDMA or GSDMA-CT had no effect on cell viability. Therefore, SpeB-cleaved GSDMA-NT induces pyroptosis only within cells.
GSDMA-NT disrupts acidic lipid membranes
To better understand how GSDMA-NT causes pyroptosis, the researchers utilized GSDMA fragments containing residues 1-214 (GSDMA1-214) or 1-246 (GSDMA1-246) markers. Protein-lipid coverage assays revealed that GSDMA1-246 binds tightly to phosphatidylserine (PS), 3-O-sulfogalactosylceramide, and cardiolipin (CL). Full-length GSDMA and GSDMA1-214 do not bind to any lipids on the band, whereas GSDMA1-246 binds phosphatidylethanolamine (PE)-phosphatidylcholine (PC) liposomes containing PS or CL, but not Binds to liposomes containing only PE and PC. When incubated with crosslinkers or liposomes, full-length GSDMA exists as a monomer, while GSDMA1-246 forms oligomers. In contrast to mSpeB, GSDMA incubated with SpeB permeated PS- or CL-containing liposomes in a concentration-dependent manner. Concomitantly recombinant GSDMA1-246 resulted in liposome leakage, whereas full-length GSDMA or GSDMA-CT did not. Thus, GSDMA1-246 produced by SpeB cleavage binds and disrupts acidic lipid membranes.
Deletion of 1Gsdma1 that disrupts acidic lipid membranes attenuates anti-GAS immunity
The mouse genome contains three GSDMA homologues, Gsdma1, Gsdma2, and Gsdma3; among them, Gsdma1 and Gsdma3 are selectively expressed on the skin. Co-expression of Gsdma1 with SpeB generates Gsdma1-NT, while SpeB does not cleave Gsdma3. In the presence of SpeB, WT Gsdmal1, can trigger leakage of PS and CL liposomes, leading to pyroptosis. Electroporation of recombinant SpeB into covR/S-mutant strain-infected keratinocytes did not induce pyroptosis as in WT infection, confirming that Gsdma1 is a substrate of SpeB that induces pyroptosis in mice.
Next, we assessed the in vivo role of GSDMA in host defense against GAS infection. Unlike WT mice, Gsdma1-/--infected mice did not have severe skin necrosis lesions, with intact epithelium and few infiltrating neutrophils (Fig. 4c–e). Gsdma1 cleavage was detected in skin lesions of WT GAS-infected mice, but not in ΔspeB-infected ones (Fig. 4f). GAS was observed in Gsdma1-/--infected mouse keratinocytes, and the mRNA levels of Il6, Ccl2, and Ccl5 were significantly reduced (Fig. 4g), but Gsdma1-/--infected mice were prone to more severe systemic infection, Spleen and liver had several log higher bacterial loads (Fig. 4h). More than half of Gsdma1-/--infected mice died within 5 days, while the majority (15/18) of WT-infected mice survived (Fig. 4i). Gsdma1-/--infected mice did not develop necrotic skin lesions even at higher concentrations of GAS infection. In addition, no covR/S mutation or other regulatory gene mutation was detected in the spleen bacteria of the two groups, excluding the role of potential mutations in bacterial transmission or systemic pathogenesis. Notably, WT and Gsdma1-/- mice were equally susceptible to subcutaneous ΔspeB GAS infection, subcutaneous S. aureus infection, or intraperitoneal WT GAS infection (Fig. 4c–e, g–i, j, k). Therefore, SpeB-mediated cleavage of GSDMA has an important role in host defense against GAS M1T1 infection.

Figure 4 Deletion of Gsdma1 attenuates anti-GAS immunity
In this study, we found that GSDMA acts as both a sensor for sensing GAS SpeB exotoxin invasion and an effector for epithelial cell pyroptosis, adding a novel mechanism for host immune recognition and response to pathogenic microbial infection. Through the pyroptosis reaction of host cells, SpeB toxin can be converted into bacterial invasion and adhesion, reducing the risk of systemic infection. At the same time, SpeB is very important for the colonization of GAS and competition with other microorganisms for colonization. Since GSDMA's broad protection in mammals improves mammals' ability to resist systemic infection, it will be interesting to investigate whether people with genetic variants in GSDMA are more susceptible to GAS infection.




Kommentare